
The landscape of nucleic acid sequencing is continuously evolving to meet research challenges and address limitations. Today, two main methods are available: short-read and long-read sequencing.
What is Short-Read Sequencing
Short-read sequencing is currently the most widely used approach, with read lengths usually ranging from 50 to 300 base pairs. Although multiple new sequencing technologies have surfaced and been introduced to the market, the most common method for short-read sequencing is Illumina’s sequencing by synthesis (SBS).
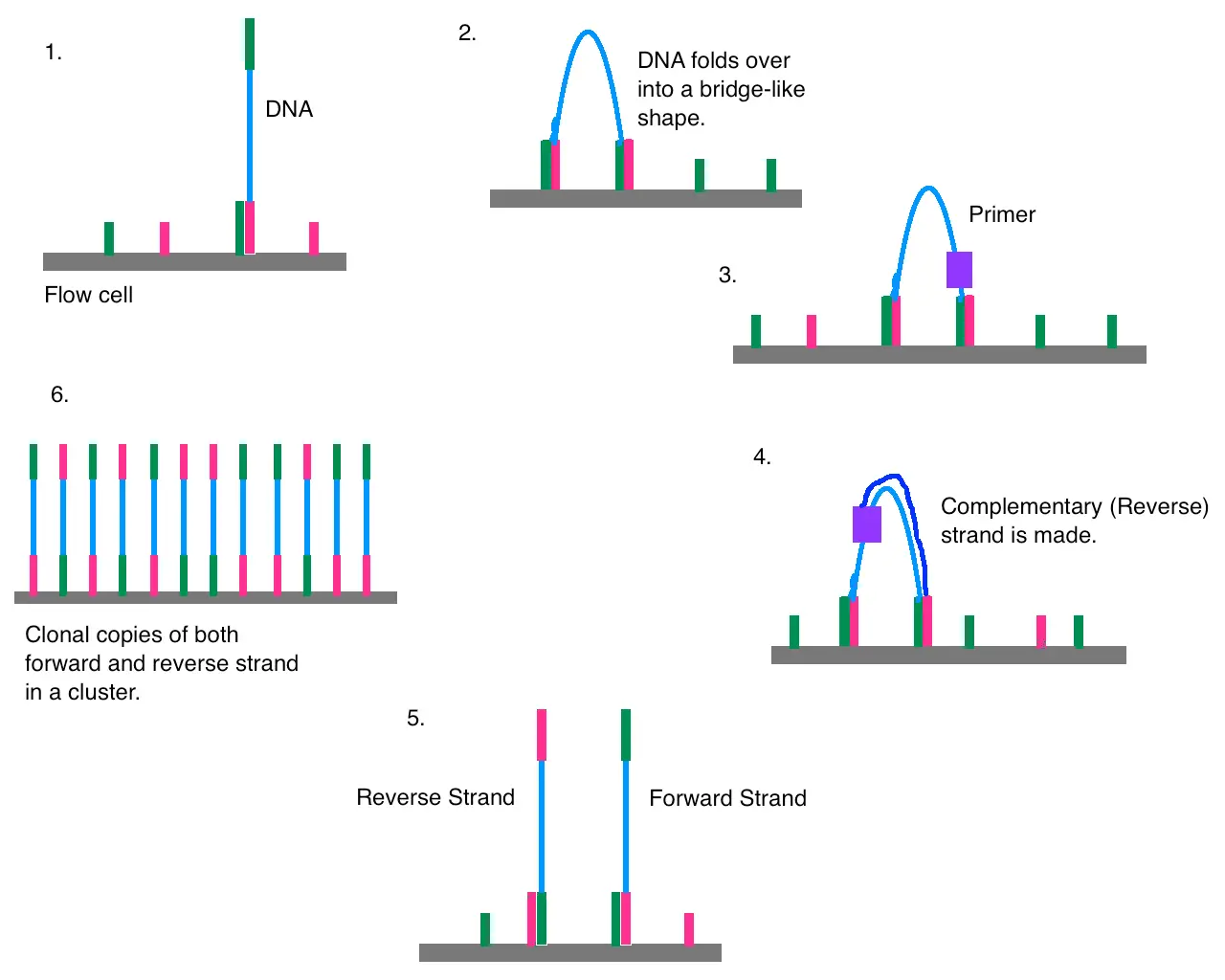
Figure 1. Illustrates the principle of the Illumina sequencing by synthesis technology in 5 steps. Sequencing by synthesis (SBS) anneals adapterized fragments to a solid support in the flow cell, a reaction vessel for sequencing, and then converts them to a single stranded template. A primer anneals to the template, and then a polymerase incorporates complementary nucleotides. The incorporation of each nucleotide is detected to sequence the template strand.
Regardless of what short-read sequencing technology is utilized – SBS or other – the number of reads range from millions to hundreds of billions depending on the sequencer. This means researchers can get higher number of reads that align to a reference genome or a subset of the genome. This alignment to the region of interest is known as coverage. High coverage enables high-confidence SNP and mutation calling. Bioinformatics pipelines are then used to assemble these random fragments into a continuous sequence.
What is Long-Read Sequencing
Long-read sequencing is a technique that can sequence long strands of DNA or RNA in one go, without breaking it up into smaller fragments. Anywhere from a few thousand to hundreds of thousands of bases can be sequenced by generating individual reads. Two primary examples of long-read sequencing technologies are Oxford Nanopore sequencing and Pacific Biosciences (PacBio) Single Molecule Real-Time (SMRT) sequencing.
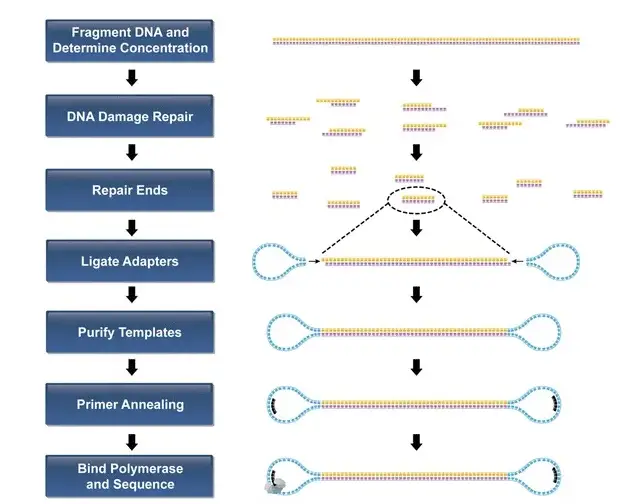
Figure 2. The PacBio (Pacific Biosciences) sequencing technique is the most widely used third generation sequencing method. PacBio Template Preparation Workflow builds a library before proceeding with the actual sequencing. For the construction of the library it is necessary to fragment the extracted DNA to be sequenced, generally by sonication and, subsequently, to add circular adapters to the ends of the DNA fragments obtained. These are called SMRTbell DNA. After building the library samples proceed directly with sequencing without going through amplification steps.
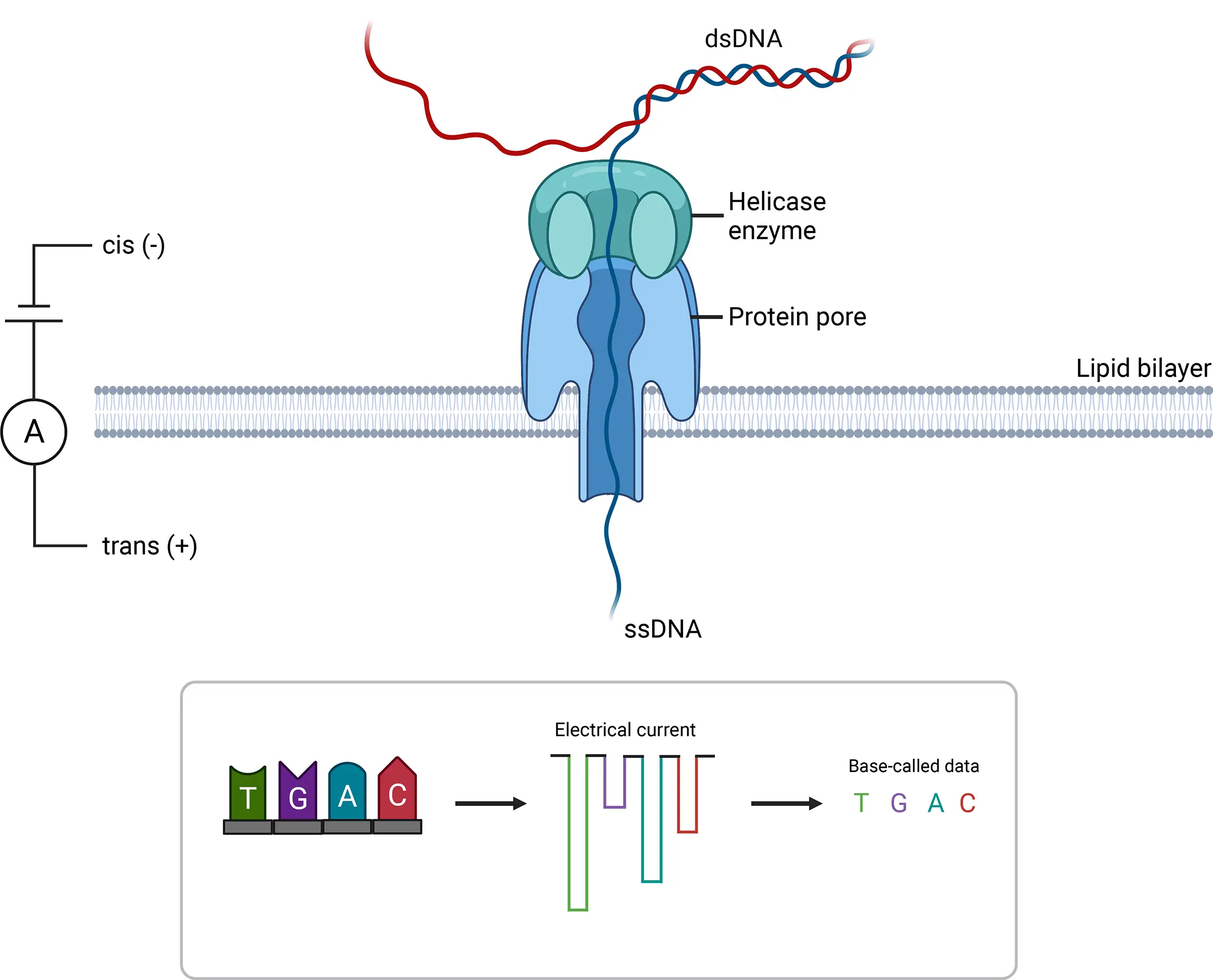
Figure 3. Oxford Nanopore sequencing is a key technology that passes single-stranded DNA or RNA molecules through nanopores embedded in a membrane. As the molecules move through the nanopore, changes in the ionic current are measured and interpreted as nucleotide sequences.Credit: Beckett et al 2021.1
Short-Read vs Long-Read Sequencing
Advantages of short-read sequencing:
- Short-read sequencing is cost-effective as it is less expensive than both traditional methods and long-read sequencing.
- Short-read sequencing is very accurate with a base-calling accuracy of over 99.9%.
- The DNA input amount and length for this technology are flexible with various NGS library preparation solutions available for ultra-low inputs or fragmented samples.
However, short-read sequencing is not without a few disadvantages.
Limitations of short-read sequencing:
- The need for a multi-step library preparation process increases the time required for experimental set-up and execution.
- Failure to generate adequate overlap between the short genomic fragments can lead to gaps in the data.
- It can be difficult to resolve and accurately characterize repetitive regions.
Advantages of long-read sequencing:
- Long-read sequencing is capable of recognizing structural variation in the genome such as large insertions/deletions, inversions, repeats, duplications, and translocations.
- This sequencing technology can also be used to phase SNPs into haplotypes – the process of using statistical methods to infer the haplotypes that are inherited from each parent based on single nucleotide polymorphism (SNP) data.
- Long-read sequencing can resolve splicing events in full-length cDNA.
While long-read sequencing has several advantages for accurately reconstructing genomes or detecting complex structural variants, it also has its drawbacks in terms of cost and throughput.
Limitations of long-read sequencing:
- Long-read sequencing can often be more expensive per base compared to short-read methods, though costs are decreasing as the technologies mature.
- Long-read methods have higher error rates compared with short-read sequencing.
- Long-read sequencing generally produces fewer reads compared to short-read technologies, thus limiting its utility for high-throughput applications requiring high coverage. High coverage sequencing is a process that sequences a region of genome multiple times to increase the confidence in the resulting data. Less reads results in lower coverage.
A Comprehensive Portfolio of NGS Preparation Methods
NGS Library Preparation methods offered by Zymo Research utilize various approaches to prepare nucleic acids for sequencing. The portfolio of solutions offered has been optimized with virtually no limit on the sample source from which the nucleic acids are extracted, or the species. Understanding that every sequencing project is unique, requiring a tailored approach to work within the limitations of the sample, , Zymo has provided solutions of products and NGS services. Whether it’s low input, compromised nucleic acids integrity, or unique sample sources, the Zymo-Seq portfolio helps leveraging the power of Next-Generation Sequencing to propel the researcher’s project forward.
Citations
- Angela H. Beckett, Kate F. Cook, Samuel C. Robson; A pandemic in the age of next-generation sequencing. Biochem (Lond) 23 December 2021; 43 (6): 10–15. doi: https://doi.org/10.1042/bio_2021_187.