Evaluating Quality of Input RNA for NGS Library Preparation
RNA-sequencing (RNA-Seq) has become an increasingly popular technique for transcriptome profiling[1]. The preparation of high-quality RNA-Seq libraries is critical to the reliability of the sequencing data, in which the integrity and purity of the input RNA play a critical role. Therefore, it is a good practice to perform quality control on the input RNA before proceeding to library preparation. Here, we provide some top recommendations to help preserve and verify the quality of input RNA for more robust RNA-Seq libraries.
About RNA Extraction
Degraded RNA can bias measurements of gene expression, provide uneven gene coverage, and prevent differentiation between alternatively spliced transcripts[2]. Thus, it is recommended to use high-integrity RNA as input whenever possible. When processing RNA samples:
- Extract RNA from samples immediately upon collection to reduce cellular RNase activity whenever possible. If temporary sample storage is needed, preserve the samples in a stabilization solution such as DNA/RNA Shield or TRIzol®, as the lytic properties of these products deactivate cellular RNases effectively.
- Use sterile filter tips during the entire workflow to minimize RNase and cross-sample contamination.
- Keep purified RNA on ice while in use and store at -80°C in aliquots to reduce freeze-thaw cycles.
- Treat RNA samples with DNase I to eliminate contaminating DNA. The presence of DNA may introduce bias in the bioinformatic processing and data analysis of the resulting library.
Assess the Quality of Input RNA for Reliable NGS Library Preparation
Perform Quality Control (QC) on your input RNA:
- Characterize the integrity of the RNA by obtaining fragment sizes and RNA Integrity Number (RIN). High quality RNA will have two prominent, slow-migrating bands representing the rRNA fragments (e.g., 18S and 28S for HeLa Cells). Smearing and a prominence of smaller fragments indicates RNA degradation.
- Determine the fragment size and RIN by a preferred method such as Agilent’s TapeStation if available (example data shown in Figure 1).
- Alternatively, using traditional gel electrophoresis can also provide the size distribution information in a similar manner (image similar to Figure 1A).
- Evaluate the purity of the input RNA using a UV-Vis spectrophotometer such as a NanoDrop™ to ensure the RNA is free from contaminating phenol and chaotropic salts. For pure RNA, the ideal A260/A280 ratio is around 2.0, and ideal A260/A230 around 2.0-2.2.
- Small changes in pH and shift in wavelength accuracy as small as 1 nm could result in ~ 0.2 change in the A260/A280 ratio[3]. Thus we recommend using the same buffer to elute RNA and characterize them on the same spectrophotometer if comparing RNA from multiple samples or conditions.
- Additionally, be aware of the level of contamination in your sample when using NanoDrop™ for quantification, since free nucleotides and contaminants can bias the data.[4,5]
- Therefore, we recommend quantifying RNA using a more specific method, such as Qubit® fluorometric quantification, which uses dyes specific for DNA or RNA.
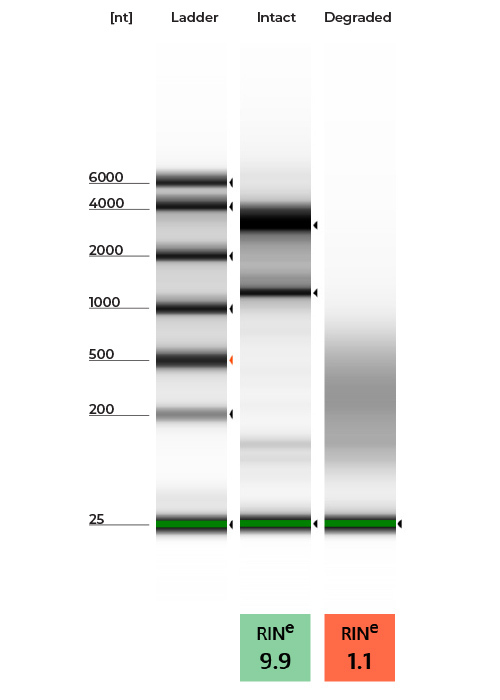
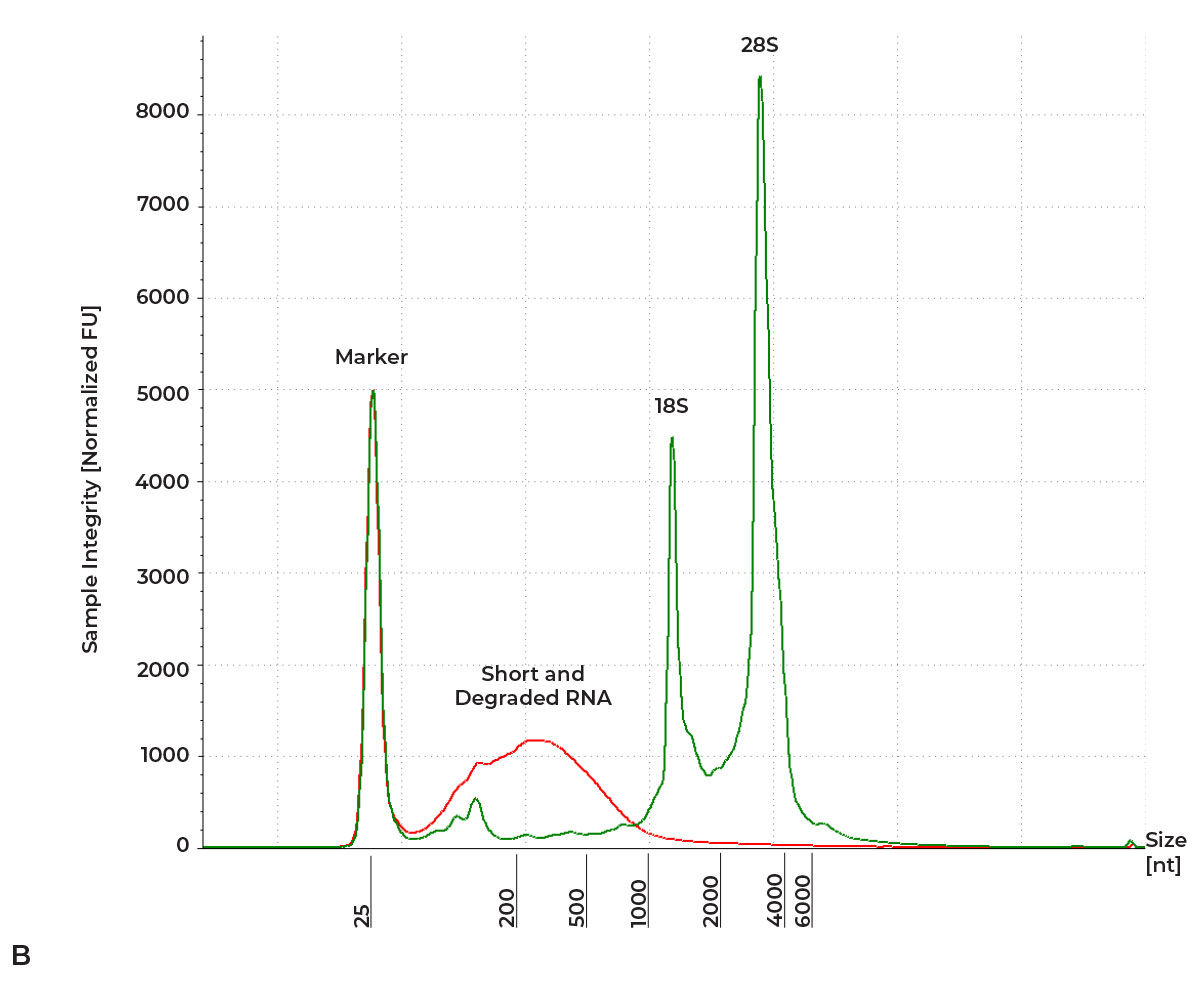
Figure 1: A) High Quality RNA vs. Degraded RNA Gel Image. B) High Quality RNA vs. Degraded RNA Electropherogram. High quality and intact RNA is indicated by the presence of two slow-moving bands (Green). In contrast, degraded RNA is indicated by an accumulation of short, fast-moving fragments (Red). RNA characterized on an RNA ScreenTape with 4150 TapeStation System.
Following these tips and tricks can help you prepare RNA samples that are suitable for RNA-Seq and produce reliable sequencing data.
Learn More About Zymo's RNA NGS Library Prep product
Learn MoreReferences
- Hong, M.; Tao, S.; Zhang, L.; Diao, L. T.; Huang, X.; Huang, S.; Xie, S. J.; Xiao, Z. D.; Zhang, H. RNA sequencing: new technologies and applications in cancer research. J Hematol Oncol 2020, 13 (1), 166. DOI: 10.1186/s13045-020-01005-x.
- Kukurba, K. R.; Montgomery, S. B. RNA Sequencing and Analysis. Cold Spring Harb Protoc 2015, 2015 (11), 951-969. DOI: 10.1101/pdb. top084970.
- Wilfinger, W. W.; Mackey, K.; Chomczynski, P. Effect of pH and ionic strength on the spectrophotometric assessment of nucleic acid purity. Biotechniques 1997, 22 (3), 474-476, 478-481. DOI: 10.2144/97223st01.
- Unger, C.; Lokmer, N.; Lehmann, D.; Axmann, I. M. Detection of phenol contamination in RNA samples and its impact on qRT-PCR results. Anal Biochem 2019, 571, 49-52. DOI: 10.1016/j.ab.2019.02.002.
- Shen, C.-H. Detection and Analysis of Nucleic Acids. In Diagnostic Molecular Biology, Chang-Hui, S. Ed.; Academic Press, 2019; pp 167-185.





